![]()
![]()
![]()

Note: this package is in early stages of development. (manuscript in prep)
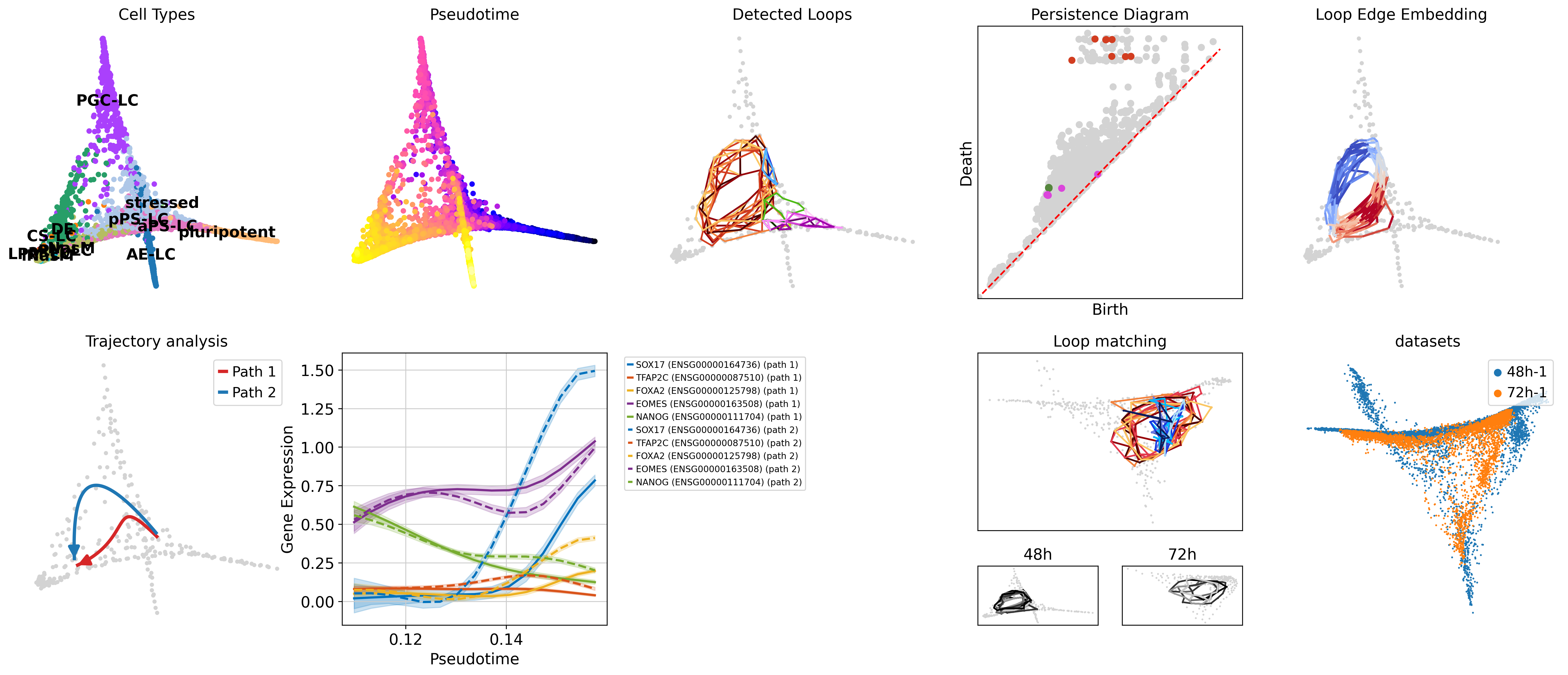
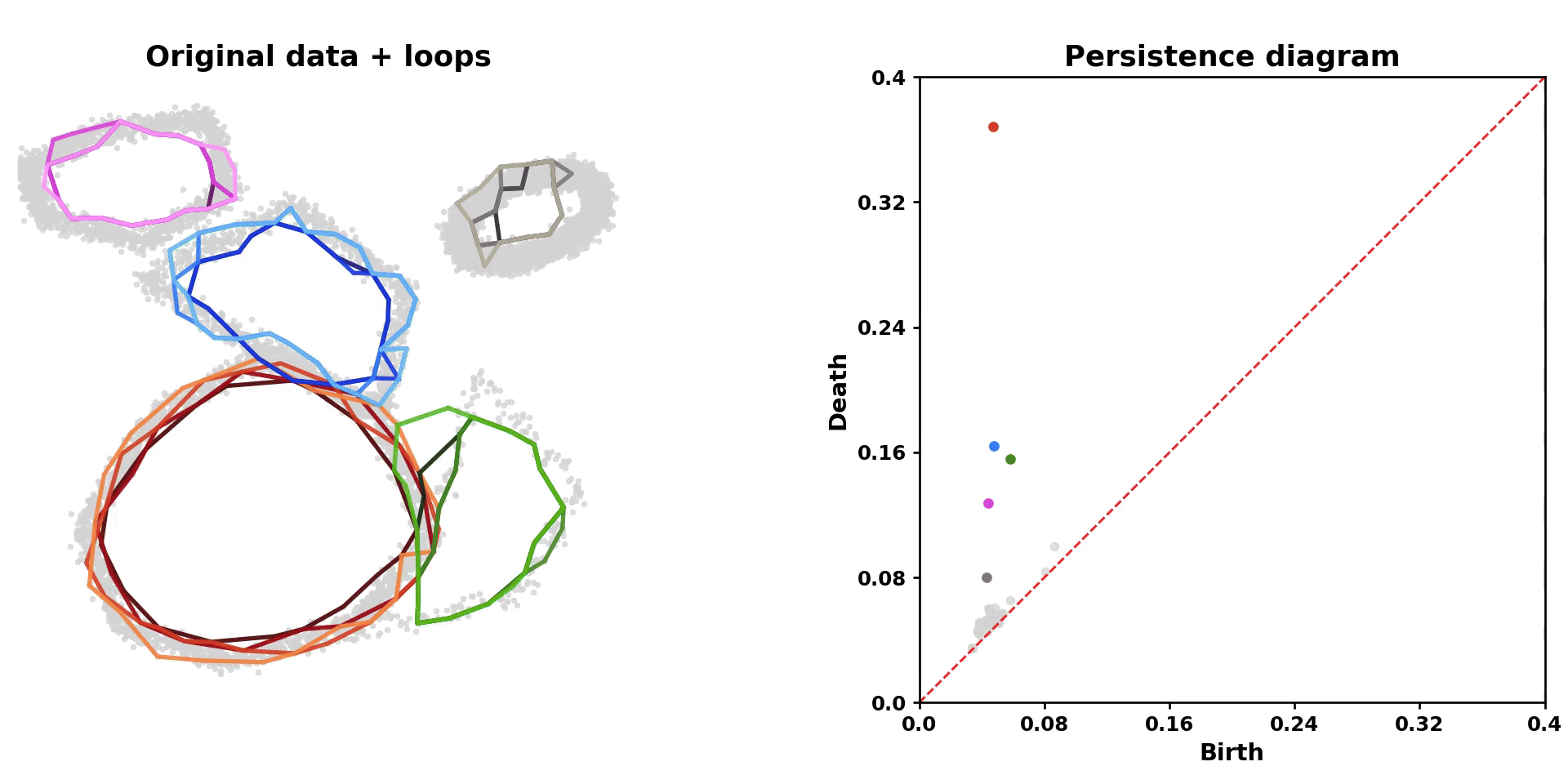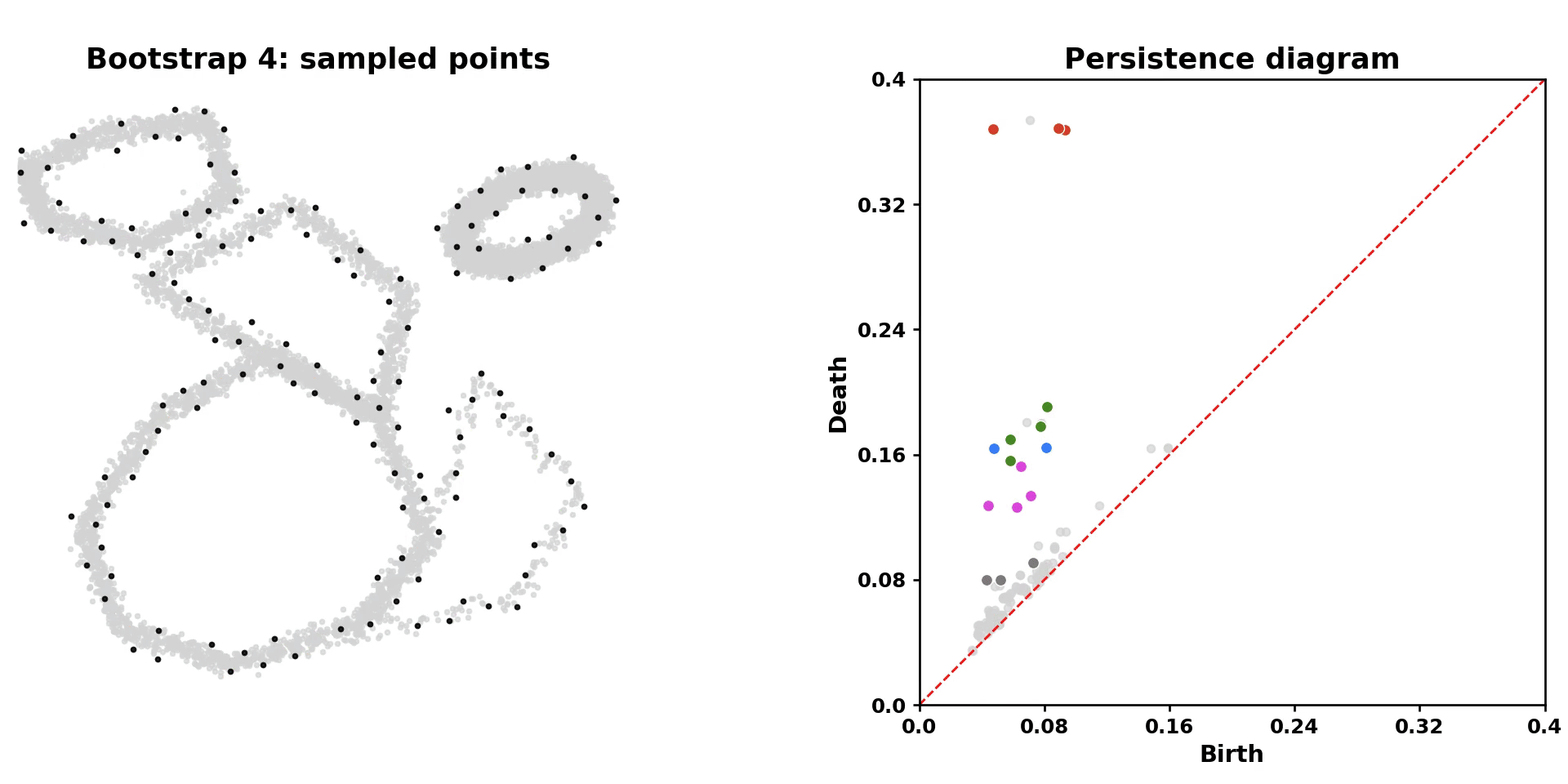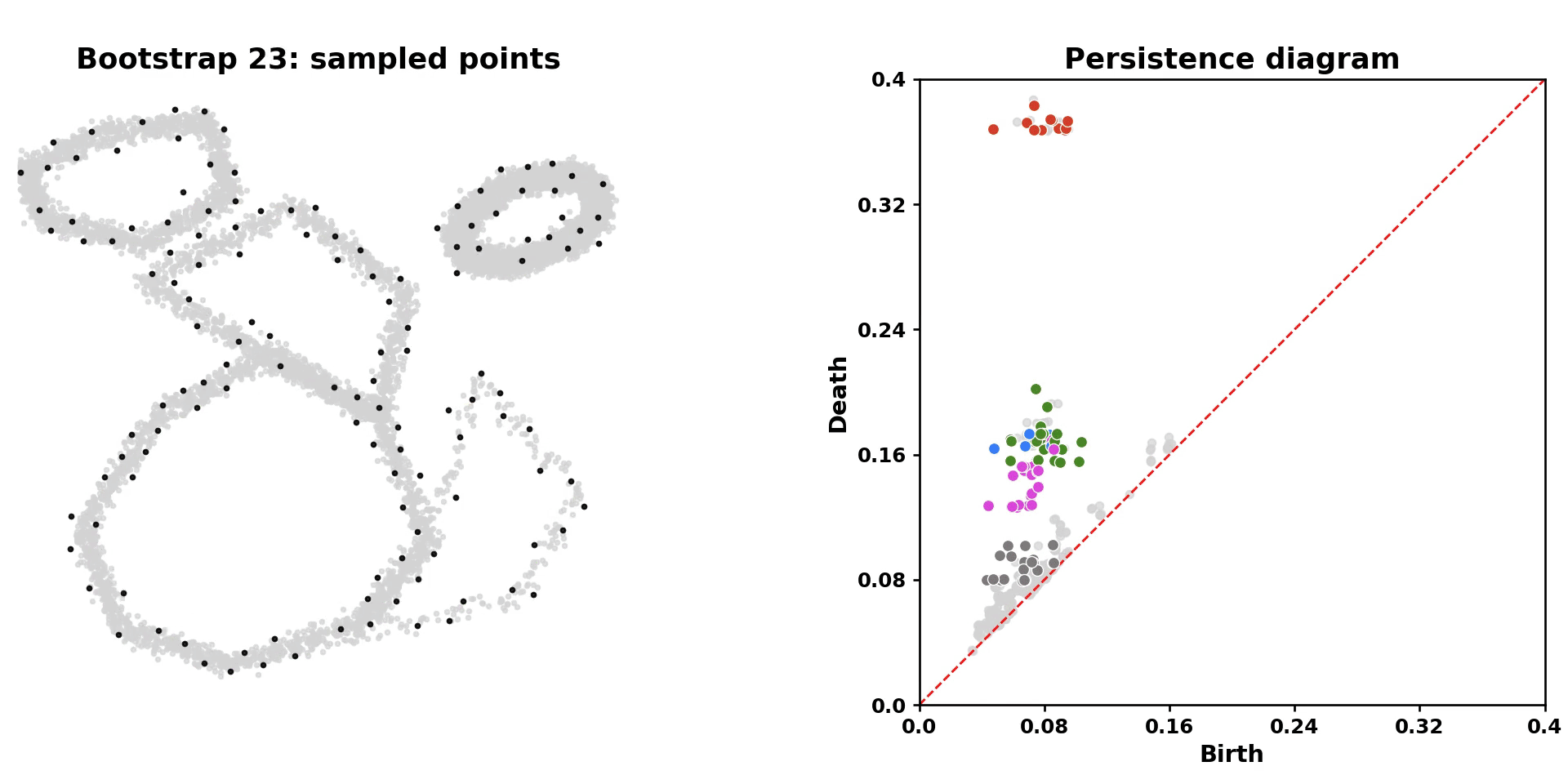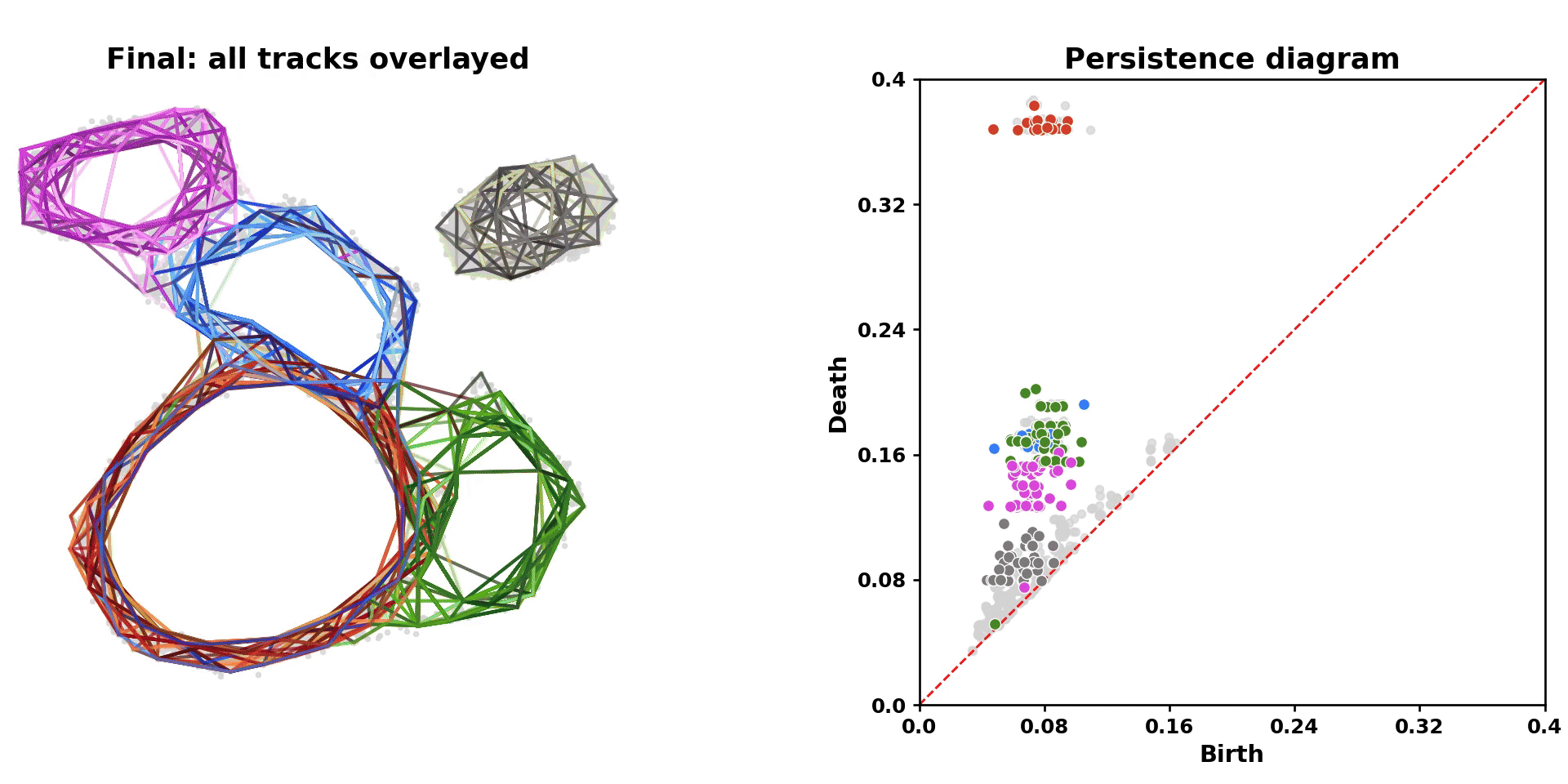
scLoop is a library to identify statistically significant loops in single-cell RNA-seq data. In brief, it bootstraps the data points and matches loops across the bootstrapped samples through both image homology and topological equivalence, which assigns confidence to individual loops in the dataset. In addition, it supports cross datasets loop matching. The core computation routine is compiled and parallelized.
|
|

|
|
import scloop as scl
# Preprocess anndata
scl.pp.prepare_adata(adata, downsample=True, n_downsample=500)
# Identify statistically significant loops
scl.tl.find_loops(adata, ...)
# Compute trajectories, gene trends, and important features of each loop
scl.tl.analyze_loops(adata, ...)
# Match loops across datasets
scl.tl.match_loops([adata1, adata2, ...], ...)make buildor
make rebuildmake syncor
make full-syncto prevent partial compilation of some modules
Note: this package is in early stages of development. The current build will have issues.
pip install scloopFeel free to contribute (prs, issues, discussions, ...)
src/scloop/
├── analyzing
│ ├── bootstrap.py
│ ├── feature_selection.py
│ ├── gene_trend.py
│ ├── hodge.py
│ ├── __init__.py
│ └── stats.py
├── benchmarking
│ ├── benchmarking_slicer.R
│ ├── datasets.py
│ ├── hf_registry.yaml
│ ├── __init__.py
│ ├── install_r_packages.R
│ ├── lle_1.1.tar.gz
│ ├── renv
│ │ ├── activate.R
│ │ ├── library
│ │ ├── settings.json
│ │ └── staging
│ ├── renv.lock
│ ├── SLICER
│ │ ├── data
│ │ ├── DESCRIPTION
│ │ ├── man
│ │ ├── NAMESPACE
│ │ ├── R
│ │ ├── README.md
│ │ ├── SLICER.Rproj
│ │ └── vignettes
│ ├── slicer_results.csv
│ └── splatter
│ ├── codecov.yml
│ ├── DESCRIPTION
│ ├── index.md
│ ├── inst
│ ├── LICENSE
│ ├── man
│ ├── NAMESPACE
│ ├── NEWS.md
│ ├── pkgdown
│ ├── _pkgdown.yml
│ ├── R
│ ├── README.md
│ ├── tests
│ └── vignettes
├── computing
│ ├── boundary.py
│ ├── divergence.py
│ ├── embedding.py
│ ├── hodge_decomposition.py
│ ├── homology.py
│ ├── __init__.py
│ ├── loops.py
│ ├── matching.py
│ └── utils.py
├── data
│ ├── analysis_containers.py
│ ├── base_components.py
│ ├── boundary.py
│ ├── constants.py
│ ├── containers.py
│ ├── __init__.py
│ ├── metadata.py
│ ├── ripser.cpp
│ ├── ripser.hpp
│ ├── ripser_lib.cpp
│ ├── ripser_lib.pyx
│ ├── types.py
│ └── utils.py
├── __init__.py
├── io
│ └── __init__.py
├── matching
│ ├── cross_dataset.py
│ ├── data_modules.py
│ ├── __init__.py
│ ├── mlp.py
│ └── nf.py
├── plotting
│ ├── _cross_match.py
│ ├── custom_colormaps.py
│ ├── _hodge.py
│ ├── _homology.py
│ ├── __init__.py
│ ├── _trajectory.py
│ └── _utils.py
├── preprocessing
│ ├── delve
│ │ ├── delve.py
│ │ ├── __init__.py
│ │ └── kh.py
│ ├── downsample.py
│ ├── __init__.py
│ └── prepare.py
├── py.typed
├── tools
│ ├── _cross_match.py
│ ├── __init__.py
│ └── _loops.py
└── utils
├── denoise
│ ├── __init__.py
│ ├── Sanity
│ ├── Sanity.cpp
│ ├── Sanity_py.py
│ └── Sanity.pyx
├── distance_metrics
│ ├── discrete-frechet-distance
│ ├── frechet.cpp
│ ├── frechet_py.py
│ ├── frechet.pyx
│ └── __init__.py
├── __init__.py
├── linear_algebra_gf2
│ ├── GF2toolkit
│ ├── gf2toolkit_lib.cpp
│ ├── gf2toolkit_lib.pyx
│ ├── gf2toolkit_wrapper.cpp
│ ├── gf2toolkit_wrapper.hpp
│ ├── __init__.py
│ ├── m4ri_lib.c
│ └── m4ri_lib.pyx
├── logging.py
└── pvalues.py